|
|

|
Processed through Paypal No account required. |
Buy our over-priced crap to help pay the bills.








|
|
|
Processed through Paypal No account required. |
| File - Download Avogadro v2.0.0 | ||||||||
| Description | ||||||||
|
A plea... Deanna and I have been running this site since 2008 and lately we're seeing a big increase in users (and cost) but a decline in percentage of users who donate. Our ad-free and junkware-free download site only works if everyone chips in to offset the revenue that ads on other sites bring in. Please donate at the bottom of the page. Every little bit helps. Thank you so much. Sincerely, your Older Geeks: Randy and Deanna Always scroll to the bottom of the page for the main download link. We don't believe in fake/misleading download buttons and tricks. The link is always in the same place. Avogadro v2.0.0 👉 Advanced open source molecular editor, visualization, and computational chemistry platform. Avogadro is a powerful free and open source molecular editor and advanced visualization platform designed for computational chemistry, molecular modeling, materials science, bioinformatics, nanotechnology research, and chemistry education. It combines an intuitive interface with professional-grade tools that allow students, researchers, educators, and scientists to build, analyze, optimize, and render molecular structures in real time. 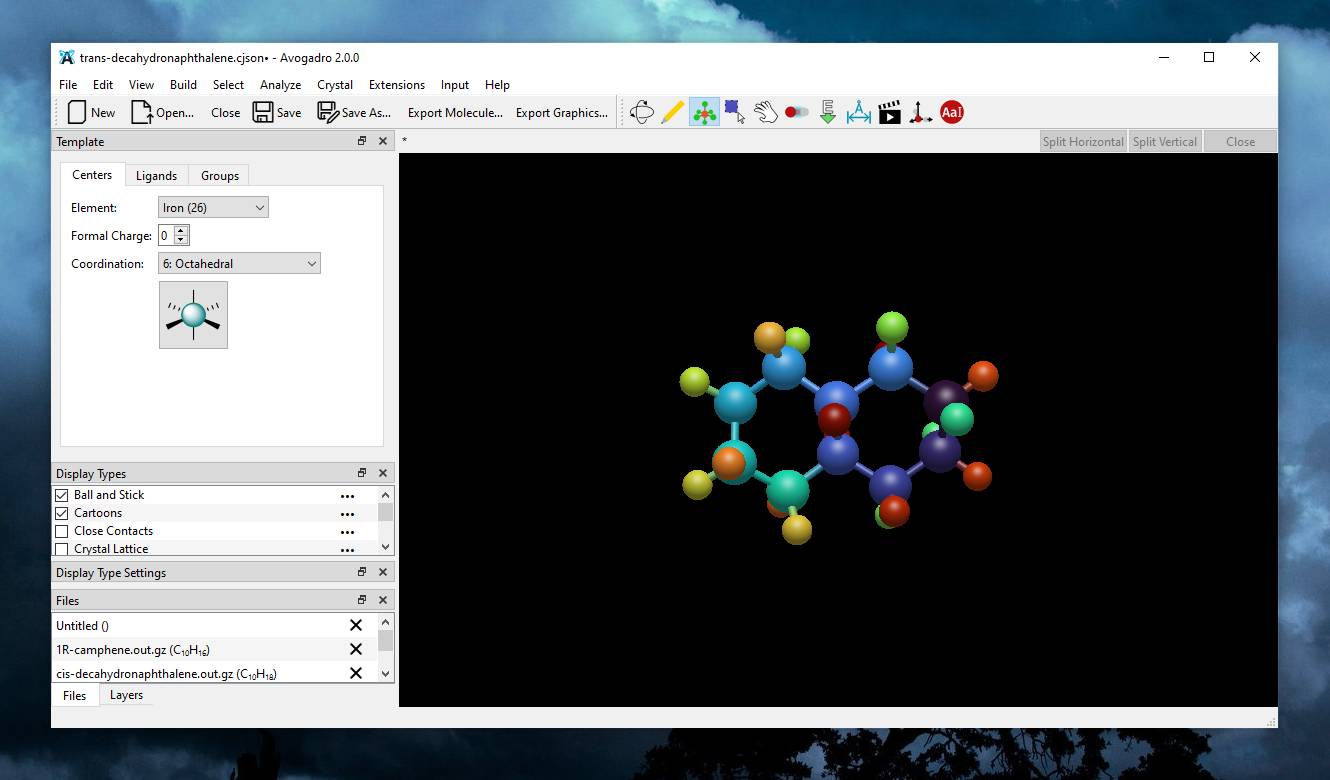 Avogadro supports everything from simple molecules to large biomolecular systems, crystalline structures, and complex materials simulations. The software includes high-quality 3D rendering, interactive molecular editing tools, force field optimization, scripting support, extensible plugins, and compatibility with a wide variety of computational chemistry packages and scientific file formats. The program can generate input files and interpret output files for many popular chemistry applications including ORCA, Gaussian, GAMESS, MOPAC, NWChem, and others. Researchers can visualize orbitals, vibrations, surfaces, electrostatic potentials, atomic properties, and molecular interactions using an interactive GPU-accelerated rendering engine. Avogadro also provides an expandable plugin ecosystem that allows users to install additional tools, file format handlers, commands, and visualization modules. Python scripting support enables automation and customization for advanced scientific workflows and educational demonstrations. 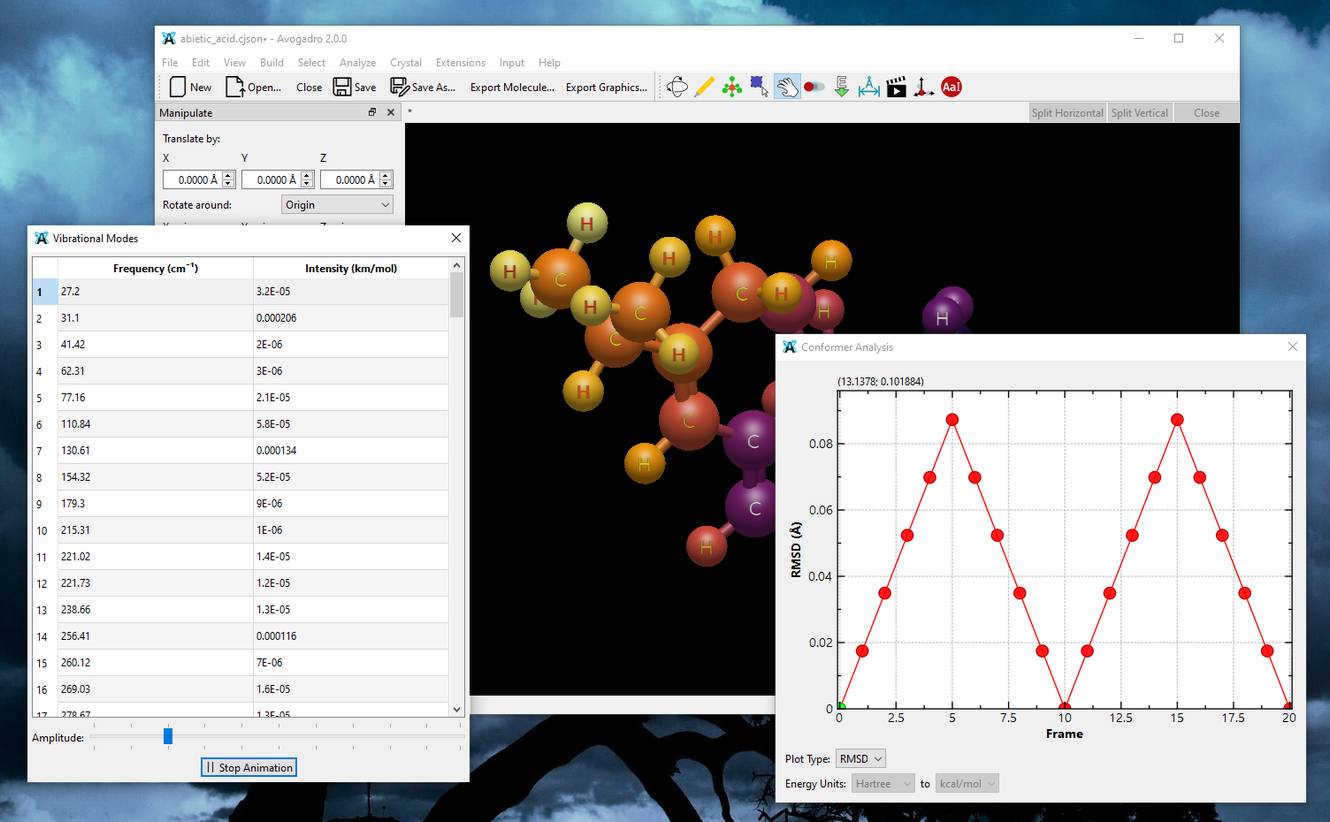 Features: • Build, edit, and manipulate molecules in an interactive 3D environment • Create organic, inorganic, crystalline, polymer, and biomolecular structures • High-quality GPU-accelerated rendering with advanced lighting and shading • Supports ambient occlusion, reflections, transparency, and visual effects • Real-time molecular geometry editing and atom placement tools • Built-in force fields for geometry optimization and structural cleanup • Interactive AutoOpt tool for fast optimization workflows • Molecular dynamics mode for simulation and structural movement studies • Visualize atomic orbitals, electrostatic potentials, vibrations, and surfaces • Generate and edit computational chemistry input files • Parse and visualize output from ORCA, Gaussian, GAMESS, MOPAC, NWChem, and more • Extensive file format support for importing and exporting molecular data • Plugin framework with downloadable extensions and community plugins • Python scripting support for automation and custom scientific workflows • Multi-threaded rendering and background processing for improved responsiveness • Syntax-highlighted ORCA input generator • Crystal and periodic structure visualization support • Educational tools suitable for chemistry classrooms and demonstrations • Open source under the BSD 3-clause license Supported operating systems: • Windows 10, Windows 11 • macOS Apple Silicon, macOS Intel • Linux • FreeBSD Supported languages: English US, English GB, English AU, English CA, Croatian, Chinese, Esperanto, French, Georgian, German, Hungarian, Japanese, Korean, Portuguese PT, Portuguese BR, Romanian, Serbian, Spanish, Tamil, Turkish, Ukrainian Changes: v2.0.0 04-01-26 New plugin system. New plugin install window and index with over 20 plugins. Faster geometry optimizations. Added faster protocol for external energy models. Added simple molecular dynamics mode to the AutoOpt tool. Improved ORCA input generator with syntax highlighting. Revised and updated documentation. Added new keyboard shortcuts. Moved AutoOpt and forcefield optimizations off the main GUI thread. Updated manage plugins interface. Implemented new plugin framework. Added write support for Molden. Many bug fixes and performance improvements. This download is for the Windows 64bit version (very bottom of page). All other download assets are below: macOS: Avogadro2-2.0.0-Darwin-arm64.dmg Avogadro2-2.0.0-Darwin.dmg Linux: Avogadro2-x86_64.AppImage Avogadro2-aarch64.AppImage Click here to visit the author's website. Continue below for the main download link. |
||||||||
| Downloads | Views | Developer | Last Update | Version | Size | Type | Rank | |
| 4,980 | 9,231 | Open Chemistry | Jun 02, 2026 - 13:13 | 2.0.0 | 115.16MB | EXE |  , out of 63 Votes. , out of 63 Votes. |
|
| File Tags | ||||||||
| molecule Avogadro editor chemistry visualization computational modeling molecular builder bioinformatics materials science quantum chemistry ORCA Gaussian GAMESS MOPAC NWChem | ||||||||
Click to Rate File Share it on Twitter → Tweet
|